


Section: New Results
Algorithms for molecular modeling and simulation
Interactive quantum chemistry
Participants : Maël Bosson, Caroline Richard, Antoine Plet, Sergei Grudinin, Stéphane Redon.
We have proposed what appears to be the first algorithm for interactive quantum chemistry simulation at the Atom Superposition and Electron Delocalization Molecular Orbital (ASED-MO) level of theory. When drawing and editing molecular systems, interactive quantum chemistry provide immediate, intuitive feedback on chemical structures. Our method is based on the divide-and-conquer (D&C) approach, which we show is accurate and efficient for this non-self-consistent semi-empirical theory. The errors induced by the D&C approach have been studied empirically and via a theoretical study of two toy models. With this method, we have demonstrated interactive quantum chemistry simulations for systems up to a few hundred atoms on a current multicore desktop computer. As the number of cores on personal computers increases, and larger and larger systems can be dealt with, we believe such interactive simulations – even at lower levels of theory – should thus prove most useful to effectively understand, design and prototype molecules, devices and materials. This result has been published in Journal of Computational chemistry [4] . Figure 7 illustrates an interactive modeling session with a benzene molecule.
|
Adaptively Restrained Particle Simulations
Participants : Svetlana Artemova, Stéphane Redon.
Particle simulations are widely used in physics, chemistry, biology [17] , [21] , and even computer graphics [13] . However, many important problems still constitute significant computational challenges, including molecular docking, protein folding, diffusion across bio-membranes, fracture in metals, ion implantation, etc. Numerous methods have been developed to accelerate particle simulations, by e.g. increasing the simulation's time step [18] , [9] , [14] , [26] , [27] , [24] , improving the computational complexity of the simulation [32] , [15] , [8] , [10] , [31] , or simplifying the system under study [19] , [29] , [28] , [16] , [11] , [31] , in particular via coarse-graining methods [20] , [33] , [30] or multiscale and multiresolution methods [25] , [22] , [23] , [12] .
We have introduced a novel, general approach to speed up particle simulations that we call Adaptively Restrained Particle Simulations (ARPS).
Our approach works by adaptively switching positional degrees of freedom on and off repeatedly during a simulation, while letting momenta evolve. The benefits of this approach are that (a) it is mathematically grounded and is able to produce stable, energy-preserving simulations; (b) it does not requires modifications to the simulated interaction potential, so that any suitable existing force-field can be directly used with ARPS; (c) under frequently-used assumptions on the interaction potential, ARPS make it possible to reduce the number of forces that have to be updated at each time step, which may significantly speed up simulations; (d) when performing constant-energy simulations, ARPS allow users to finely and continuously trade between precision and computational cost, and rapidly obtain approximate trajectories; (e) the trade-off between precision and cost may be chosen for each particle independently, so that users may arbitrarily focus ARPS on specific regions of the simulated system (e.g. a polymer in a solvent); (f) most important, when performing Adaptively Restrained Molecular Dynamics (ARMD) in the canonical (NVT) ensemble, unbiased statistics can be obtained.
We have illustrated ARPS on several numerical experiments, including a shock propagation example and a polymer-in-solvent study.
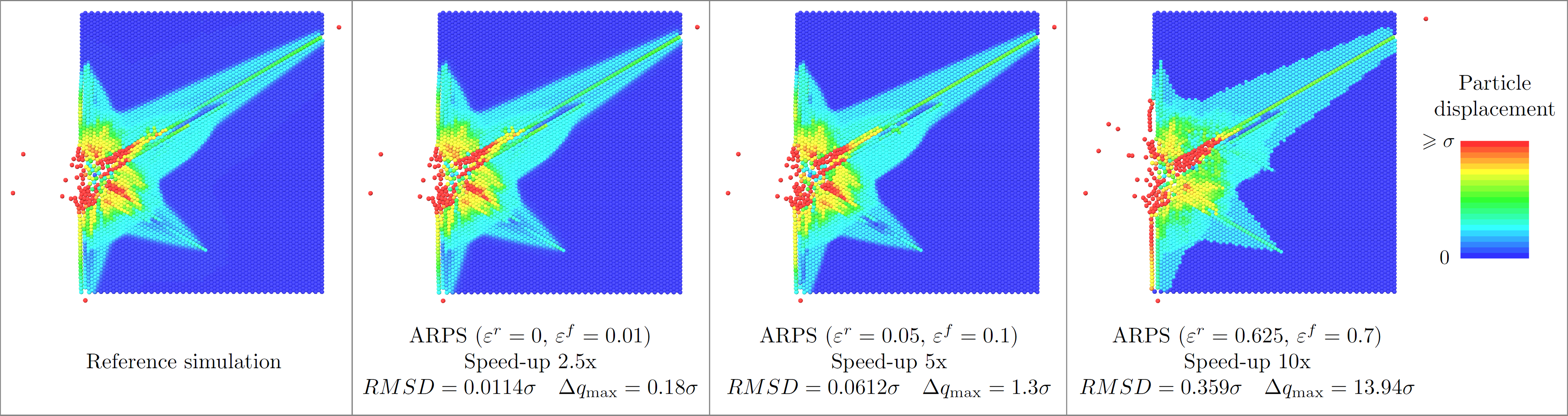
The shock propagation example demonstrates how ARPS make it possible to smoothly trade between precision and speed (Fig. 8 ).
|
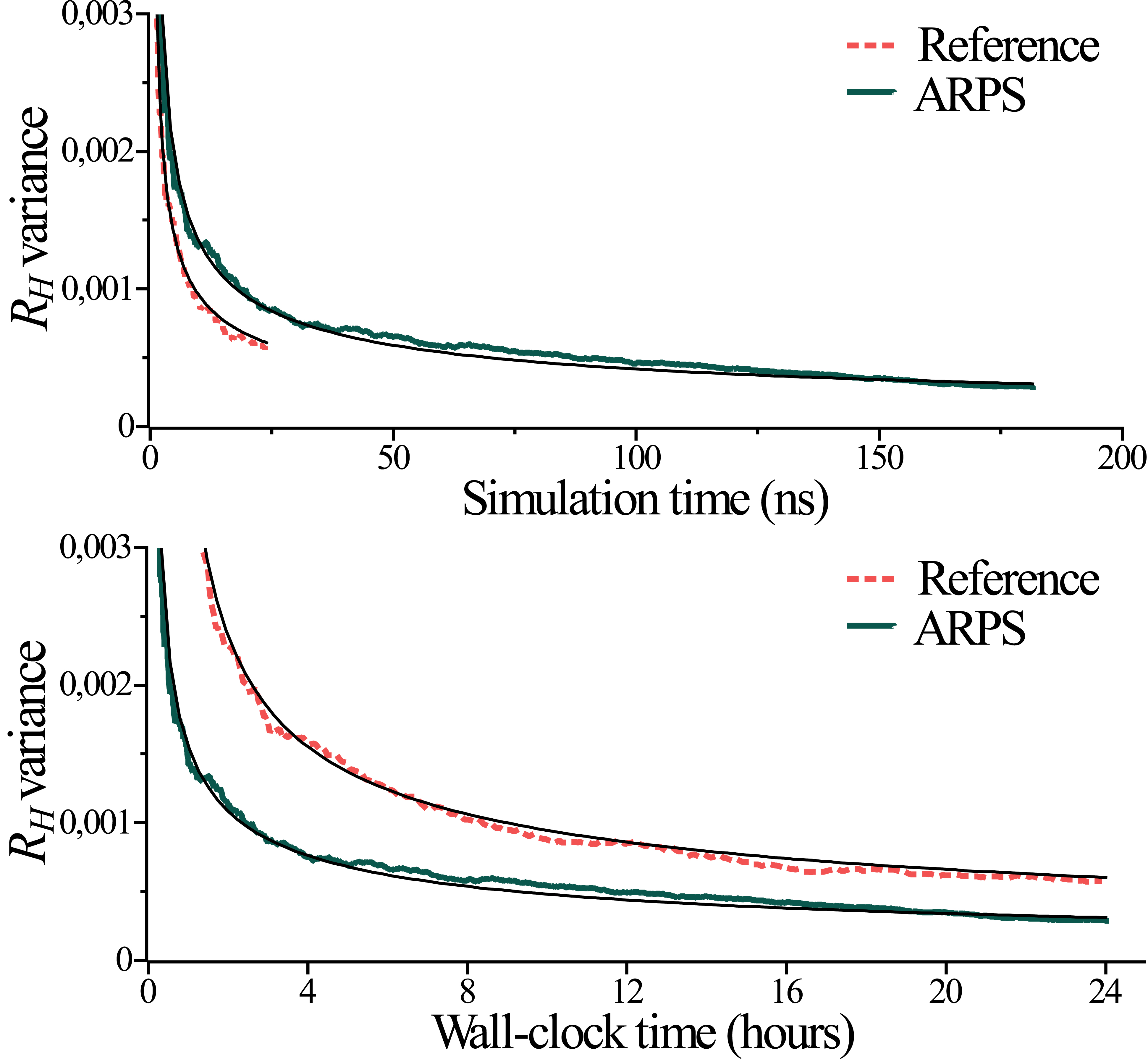
The polymer-in-solvent study shows how one may collect unbiased statistics with ARPS, and demonstrates that it can be done faster than with usual (reference) simulations, The results are shown in Fig. 9 .
|
These results have been submitted for publication.
Adaptive interactive quantum chemistry
Participants : Maël Bosson, Sergei Grudinin, Stéphane Redon.
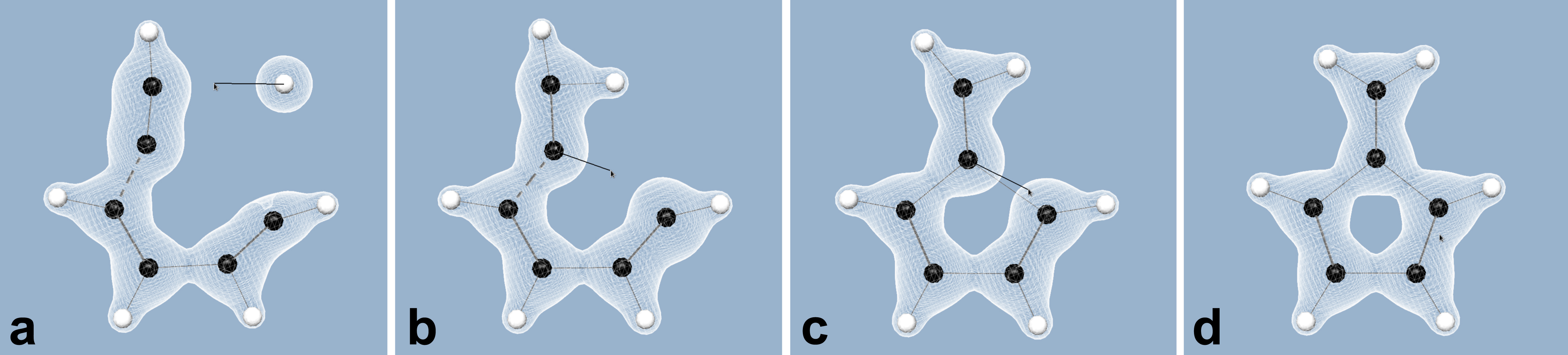
We are now working on applying the adaptive paradigm to the quantum chemistry methods, to allow for interactive editing of systems of any sizes and shapes. We are developing new methods and criteria to adaptively focus the computational resources on the most relevant parts of the system. Figure 10 illustrates our recent results in this direction. In this framework, we can already achieve interactive rates and efficient virtual prototyping for systems of size up to thousand atoms on a current desktop computer.
|
Interactive molecular modeling with haptic feedback
Participants : Aude Bolopion, Barthelemy Cagneau, Stéphane Regnier, Stéphane Redon.
In collaboration with ISIR in Paris and LISV in Versailles, we have developed a new approach for haptic interaction with molecular systems.
Molecular interactions typically have a high dynamic range (HDR), combining short-range stiff repulsive effects with long-range, soft attractive and repulsive terms. As a result, faithful haptic rendering of such molecular interactions is both important and difficult, in particular in applications where the precise perception of molecular forces is necessary (e.g. in molecular docking simulations). Traditionally, teleoperation coupling using constant gain control schemes have limited applications since they are unable to transmit to users low attractive forces without truncating repulsive ones. Furthermore, constant scaling displacement induces either instability or time-consuming experiments (displacements are slow), which deteriorates the ease of manipulation. We have described a variable gain haptic coupling method specifically designed to render high dynamic range (molecular) forces. The proposed method has been evaluated by user tests on an experiment involving two water molecules. We have observed that variable force amplification is widely appreciated, whereas variable displacement scaling is appropriate only for users that are already familiar with haptic manipulation. A complex experiment on a HIV molecule has been carried out using this variable gain system. This approach has been published in the proceedings of the 2011 World Haptics Conference [7] . Figure 11 shows SAMSON being used with a haptic interface at ISIR.
